Because mitochondria are responsible for the majority of cellular ATP synthesis, disruption in mitochondrial function can play a substantial role in DILI. Any disruption in ATP synthesis results in lower ATP levels capable of triggering disturbances in other cellular processes and ultimately leading to necrotic cell death. Parent compounds and their metabolites may disrupt ATP production in a number of ways, most commonly reduction in the mitochondrial protein gradient via uncoupling, inhibition of the electron transport chain flux, or direct inhibition of ATP synthesis. Increases in ROS/RNS production (as discussed here) can also decrease ATP synthesis.
01. Discover
What is the Mitochondrial Function Submodel?
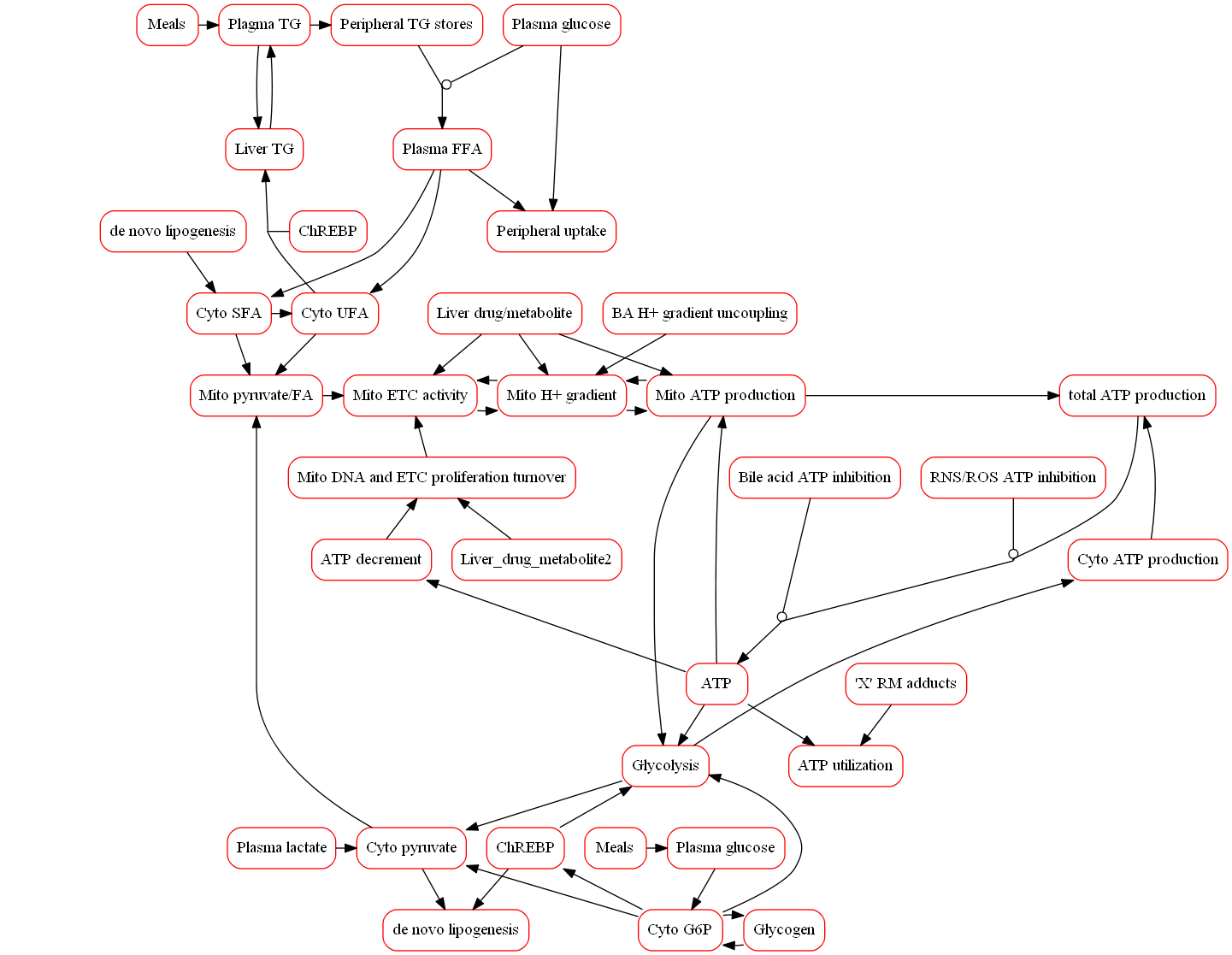
The representation of mitochondrial biochemistry within the model scope includes mitochondrial respiration, proton-motive force, and ATP production in addition to compounds that disrupt these processes. Exemplar compounds, including buprenorphine, have been simulated to refine the model and ensure that the modeled representation of mitochondrial toxicity is consistent with available data. The model also incorporates associated pathways that participate in the overall hepatic response to mitochondria toxicity in vivo such as glycolysis and tradeoffs between glucose and fatty acid oxidation. Moreover, alterations in the mitochondrial proton gradient due to the accumulation of bile acids can affect ATP synthesis and provide the ability for DILIsym to simulate how multiple mechanisms (i.e., bile acids and mitochondria) can synergistically elicit DILI.
Further, a distinct model of in vitro mitochondrial respiration and ATP production (MITOsym®) is represented and provides direct comparisons with in vitro mitochondrial assays. Cellular parameters such as oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) are used in MITOsym to determine changes in ETC activity and glycolytic function.
02. Submodels
Learn more about other submodels within DILIsym®
DILIsym is Quantitative Systems Toxicology (QST) software designed to be used during drug development to provide an indication of the potential drug-induced liver injury (DILI) hazard posed by individual molecules and/or to provide deeper insight into the mechanisms responsible for observed DILI responses at various stages of the development process.
03. Resources
Peer-reviewed Publications
